Unravelling the pathophysiology of chronic stroke lesions could yield treatments for stroke-related dementia
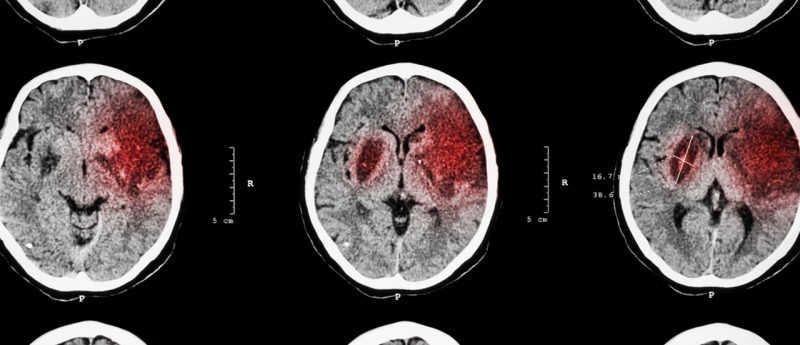
Worldwide, approximately 10 million individuals survive ischemic stroke each year (1) and although the pathophysiology of stroke injury in the brain during the acute stage is well defined, much less is known about the pathophysiology of the chronic stage. In addition to this, relatively little is understood about the mechanisms by which recovery occurs, and there are no FDA-approved therapies currently available to improve patients’ recovery following stroke. Furthermore, more than one-third of stroke survivors develop new onset dementia after stroke, the causes of which are still unclear. There is therefore a pressing need for the development of treatments for...