The neuroscience of epilepsy: inflammatory signaling as a catalyst of seizure activity
Epilepsy is a central nervous system (CNS) disorder, that can be genetically inherited or developed from neurological injury. The disorder is characterized by episodes of abnormal electrical discharges in the brain that result in abnormal motor and sensory activity; these episodes are known as seizures. Depending upon the type of seizure and area of the brain affected, the motor and sensory presentation of the seizure can vary. The underlying causal mechanisms behind the development of seizure activity are unclear, but new research suggests a vascular, immune-inflammatory role in the development of seizure activity. What does this say about the future of epilepsy treatments and the potential for preventive, rather than palliative interventions?
Seizure classification
There are many different types of seizures, but for simplicity, this article will focus on the heritable forms of generalized epilepsy and focal temporal lobe epilepsy, as they are the most common presentations. Generalized epilepsy (GE) often presents with grand mal (GM) seizures, also known as tonic-clonic seizures (TC), that involve abnormal electrical activity throughout the entire brain. This can result in loss of consciousness, generalized bodily convulsions involving alternating muscle laxity and rigidity, abnormal facial movements, and loss of bladder or bowel control. Temporal lobe epilepsy (TLE) is a focal seizure disorder, meaning that it typically occurs in the temporal lobe region of the brain. However, it is not unheard of for TLE seizure activity to progress into other brain regions to become bilateral, generalized seizures. While these seizures are focal, the seizure activity can affect cognition and sensory or motor control.
Drug-resistant epilepsy: an ongoing public health issue
Anti-convulsant therapies such as Depakote (valproic acid) and Keppra (levetiracetam), have traditionally focused on mechanisms that increase the neurotransmitter gamma aminobutyric acid (GABA) at the post synaptic terminals, while also stabilizing sodium channels to reduce neuronal hyperexcitability. This therapeutic target has been largely successful in reducing seizures but still falls short of reducing costs as patients can still experience break-through seizure activity, and many continue medications either for many years beyond seizure freedom or for their lifetime.
Despite anti-convulsant therapy drugs, neurosurgical craniotomy, focal laser ablation and biotech modalities such as neuromodulation of the vagus nerve being made available to patients, epileptic seizures continue to be a costly public health issue. Drug resistant epilepsy can occur across all seizure types and can result in neuronal cell death, sensory or motor impairments, permanent disability and death without seizure suppression. This has spurred researchers to investigate alternative models of seizure pathology and investigate new targets for interventional treatments and preventive measures.
Neuroimmunology and seizure etiology
Research investigating epilepsy has recalibrated to introduce a systemic view of the inflammatory and immunological mechanisms underlying seizure pathology (Fig. 1). The leading hypothesis is that both peripheral and central inflammatory mechanisms disturb the blood brain barrier (BBB), allowing for the passage of leukocytes and thereby triggering neuronal hyper excitability, through the release of chemokines and interleukins (IL) via astrocytes and microglia reactivity [2, 3, 4, 5, 6, 7].
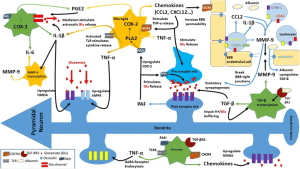
Figure 1. Inflammatory pathways [1].
These inflammatory mechanisms drive the disturbance in the BBB, ultimately leading to upregulated glutamate and electrical signaling in the hippocampus, resulting in neuronal hyper excitability. This is now thought to be a leading cause of seizure development (Fig. 1) [1, 7].
These inflammatory mechanisms however still require a catalyst, and as denoted earlier, central and peripheral inflammatory mechanisms are considered key in triggering the inflammatory molecular cascades that lead to BBB permeability, leukocyte infiltration and IL and chemokine expression. These catalysts include genetics that may result in excess inflammatory signaling and vascular injury affecting the BBB, either by the normal aging process or traumatic brain injury. CNS inflammatory mechanisms that contribute to seizure formation are especially influenced by certain genetic mutations or deletions, given that many of the known epilepsy genes that contribute to altered glial and astrocyte morphology (and TLE or GE) also fall along important metabolic pathways that regulate inflammation and cell signaling.
Familial epilepsy genes adjacent to central metabolic pathway in aging research
One such pathway is the molecular target of rapamycin (Fig. 2), an amino acid sensing serine/threonine kinase pathway. This pathway is important for cell growth and development, autophagy, mRNA proliferation, lipid synthesis and metabolism, mitochondrial metabolism and biogenesis, and vascular growth factors, all of which are vital for systemic and neuronal health, development and signal transduction [7, 8, 9].
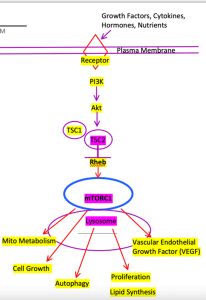
Figure 2. PI3K – TSC1/2 mTOR Pathway at Lysosome. Outputs referenced from [8].
Tuberous sclerosis is a familial epilepsy gene that often results in TLE and/or GE seizure activity and is also directly responsible for the molecular suppression of mTORC1 at Rheb, and thereby (CNS and peripheral) inflammation [7]. A homozygous or heterozygous deletion of the protein hamartin or tuberin (TSC1 or TSC2, respectively) for example, leaves mTORC1 constitutively active. This results in inflammatory signaling and altered glial and astrocyte development, which may contribute to the development of epilepsy through the aforementioned inflammatory pathways [9].
Though TSC was discovered in the 1990’s, we are just now elucidating the inflammatory signaling that may underlie the development of seizure activity from TCS2 and other familial epilepsy genes. Further, we are now seeing how the probability of an expressed or unexpressed phenotype may be based on specific variants of variable penetrance, and the added complexity of the presence of compounding upstream deletions that can be either compensatory or inflammatory [9].
Questions arise as to whether the presence of one mutation guarantees a diseased phenotype, or if the absence of an epilepsy phenotype may still present with subclinical inflammatory manifestations that contribute to both neuronal and vascular inflammatory aging over the life course, or other systemic inflammatory conditions. Could this drive BBB permeability and therefore susceptibility to seizure activity later in life? It may, especially for those aged 65 and older, which are thought to be at a higher risk for late-life seizure activity.
Systemic autoimmune and cerebrovascular implications
Since 2017, the inflammatory model has been applied to the International League Against Epilepsy (ILAE) Classification of the Epilepsies, designating autoimmune epilepsy as its own tertiary, syndromic classification as early research began to emerge [10, 11]. Evidence now suggests that epilepsy populations are known to have much higher rates of inflammatory autoimmune, cardiovascular and gastrointestinal disease; and while this provides further support for the systemic inflammatory hypothesis and that of subclinical inflammation, it also provokes questions as to whether such risk continues following remission of adolescent epilepsy [10].
Specific inflammatory mediators have been associated with different epilepsy types, as well as vascular inflammation and aging. TLE and GE have been associated with increased serum IL-1⍺, IL-1β, IL-6, TNF-a and CCL3-5 chemokines in epilepsy tissue, released by granulocytes, macrophages, lymphocytes and endothelial cells peripherally, while centrally from astrocytes and microglia, which provide evidence of the mutual importance of neuronal and vascular mechanisms in the etiological framework [5, 12].
Conversely, new research has found that late-life seizure remission may be possible through the process of endothelial senescence inherent to vascular aging. In a 20-year longitudinal study of 226 people with severe epilepsy, it was found that the inflammatory vascular damage that can mediate BBB disruption and thereby epilepsy, can also be a partially ameliorative factor in later life. Essentially, vascular damage from aging disrupts connections and networks between brain hemispheres leading to seizure cessation and drug freedom [10].
Future research
Given the vast array of literature on vascular and ‘system theories of aging’, it may therefore be beneficial for mechanisms of cellular senescence, reactive oxygen species and vascular inflammation through these mTOR kinases to be further investigated to help predict both epileptic and systemic inflammatory susceptibility through upstream or downstream associated inflammatory biomarkers [13] . Should biomarkers provide a clearer picture of the precise signaling pathways involved, this could also lead to more targeted pharmaceutical development.
1. Rana A, Musto AE. The role of inflammation in the development of epilepsy. J. Neuroinflammation, 15, 144 (2018).
2. Aronica E, Ravizza T, Zurolo E, Vezzani A. Astrocyte immune responses in epilepsy. Glia, 60(8) 1258–1268 (2012).
3. Choi J, Koh S. Role of Brain Inflammation in Epileptogenesis. Yonsei Med. J. 49(1) 1–18 (2008).
4. Choi S-H, Aid S, Choi U, Bosetti F. Cyclooxygenases-1 and -2 differentially modulate leukocyte recruitment into the inflamed brain. Pharmacogenomics J. 10(5), 448–457 (2010).
5. de Vries EE, van den Munckhof B, Braun KPJ, van Royen-Kerkhof A, de Jager W, Jansen FE. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 63, 177–190 (2016).
6. Friedman A, Dingledine R. Molecular cascades that mediate the influence of inflammation on epilepsy. Epilepsia 52(s3) 33–39 (2011).
7. Zattoni M, Mura ML, Deprez F et al. Brain Infiltration of Leukocytes Contributes to the Pathophysiology of Temporal Lobe Epilepsy. J. Neurosci. 31(11) 4037–4050 (2011).
8. Celebi-Birand ED, Karoglu ET, Doldur-Balli F, Adams MM. Chapter 11—Mammalian Target of Rapamycin (mTOR), Aging, Neuroscience, and Their Association with Aging-Related Diseases. In K. Maiese (Ed.), Molecules to Medicine with mTOR (pp. 185–203). Academic Press (2011).
9. Takei N, Nawa H. MTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neuosci. 7, 28 (2014).
10. Rajakulendran S, Belluzzo M, Novy J et al. Late-life terminal seizure freedom in drug-resistant epilepsy: “Burned-out epilepsy.” J. Neurol. Sci. 431, 120043 (2021).
11. Scheffer IE, Berkovic S, Capovilla G et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 58(4) 512–521 (2017).
12. Li G, Bauer S, Nowak M et al. Cytokines and epilepsy. Seizure 20(3) 249–256 (2011).
13. Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of Vascular Aging. Circ. Res. 123(7) 849–867 (2011).
You might also like:




